Step-by-step instruction guide
This guide will walk you through the key functionalities, enabling you to easily access and utilize the wealth of information within the riboCIRC database.
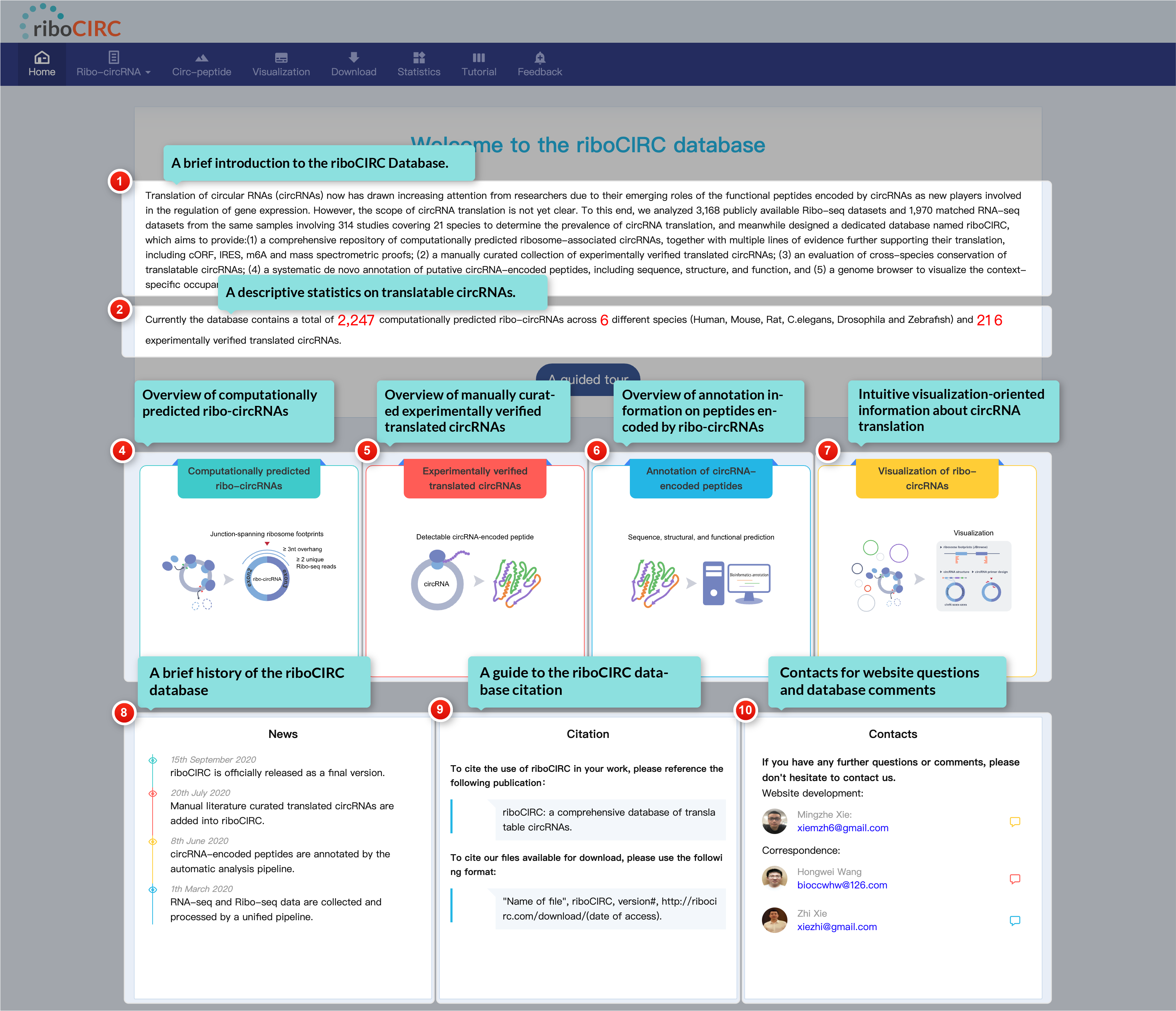
The home page serves as your gateway to riboCIRC. It features a brief introduction to the database, a summary of translatable circRNA statistics, and a Quick Start module to help you get started. You can also learn about the website's history, find citation details, and access contact information for questions or feedback. For a guided experience, click the "A Guided Tour" button to begin your online tour!
1. Computationally predicted ribo-circRNAs
Translatable circRNAs are identified through two approaches: (1) annotation-guided predictions based on known circRNAs and Ribo-seq data, and (2) context-specific predictions derived from paired RNA-seq and Ribo-seq data. Characterized translatable circRNAs include meta-information such as the new Uniform_ID, named according to "A guide to naming eukaryotic circular RNAs." Users can access detailed information, including structure and designed circRNA primer sets, by clicking on the riboCIRC_ID or the More Details button.
2. Literature-reported trans-circRNAs
This page features a manually curated encyclopedia of experimentally validated circRNAs capable of generating peptides. Unlike v1, which included literature-reported translatable circRNAs regardless of translation validity, this update applies stricter criteria. Only entries supported by multiple independent lines of evidence from the literature have been retained..
3. Cross-species conserved ribo-circRNAs
This page displays translatable circRNAs conserved across species, with conserved entries aligned in the same row for easy comparison.
The Circ-peptide page provides in silico characterization of potential peptides encoded by finite ORFs derived from ribo-circRNAs. Choosing an item from the dropdown menu reveals detailed information encompassing sequence, structure, and function. This resource establishes a valuable foundation for circRNA translation research and offers a critical starting point for exploring the biological importance of circRNA-encoded peptides.
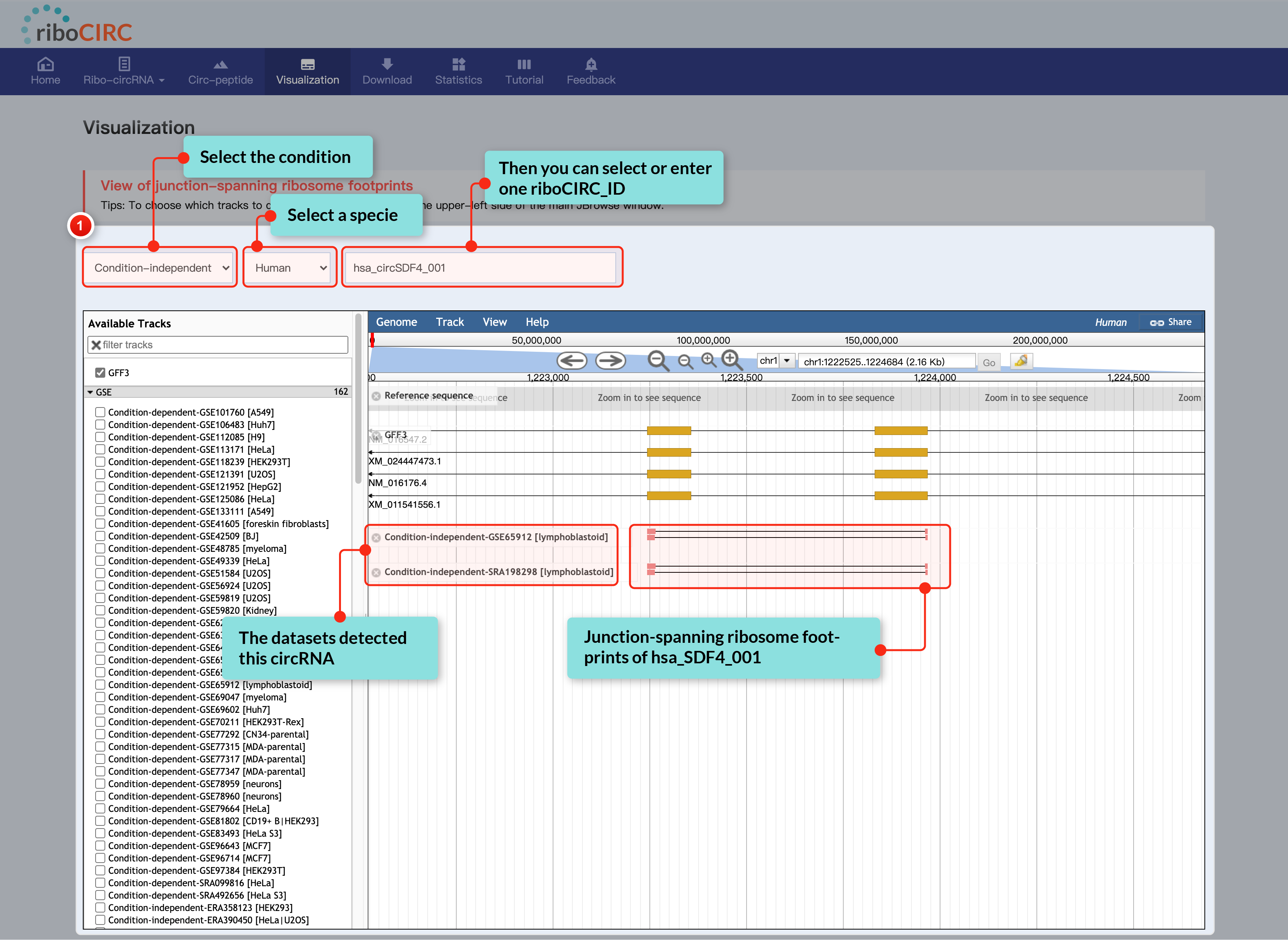
The Visualization page offers clear and interactive views of circRNA translation, highlighting junction-spanning ribosome footprints. Users can select translatable circRNAs and ribosome-profiling data tracks based on context, accessible through JBrowse's left sidebar.
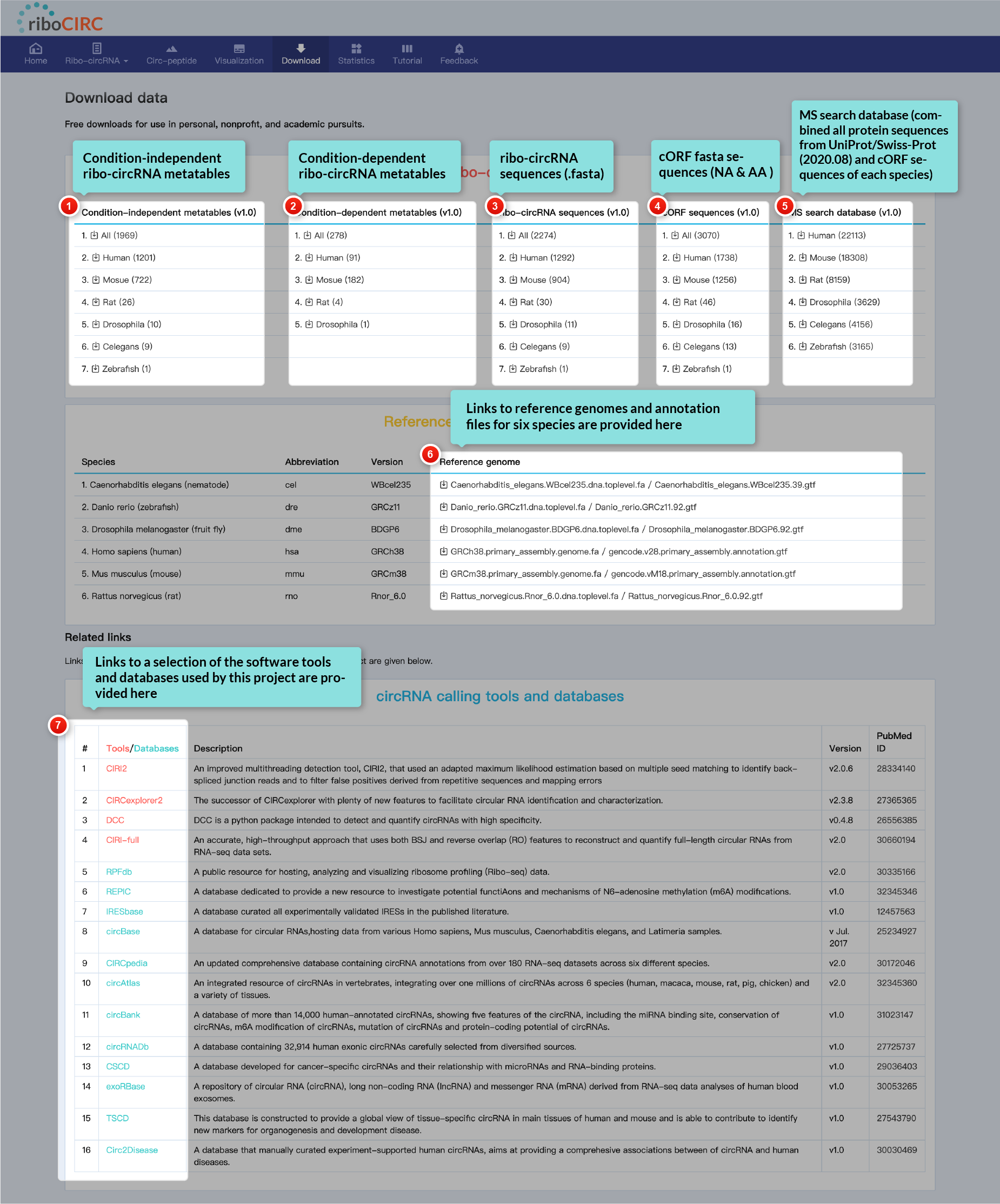
All data within this database is freely available for download. Users are welcome to utilize this data for personal, non-profit, and academic purposes without restriction.
This page provides a grand summary that aggregates data from all records currently visible in the database.
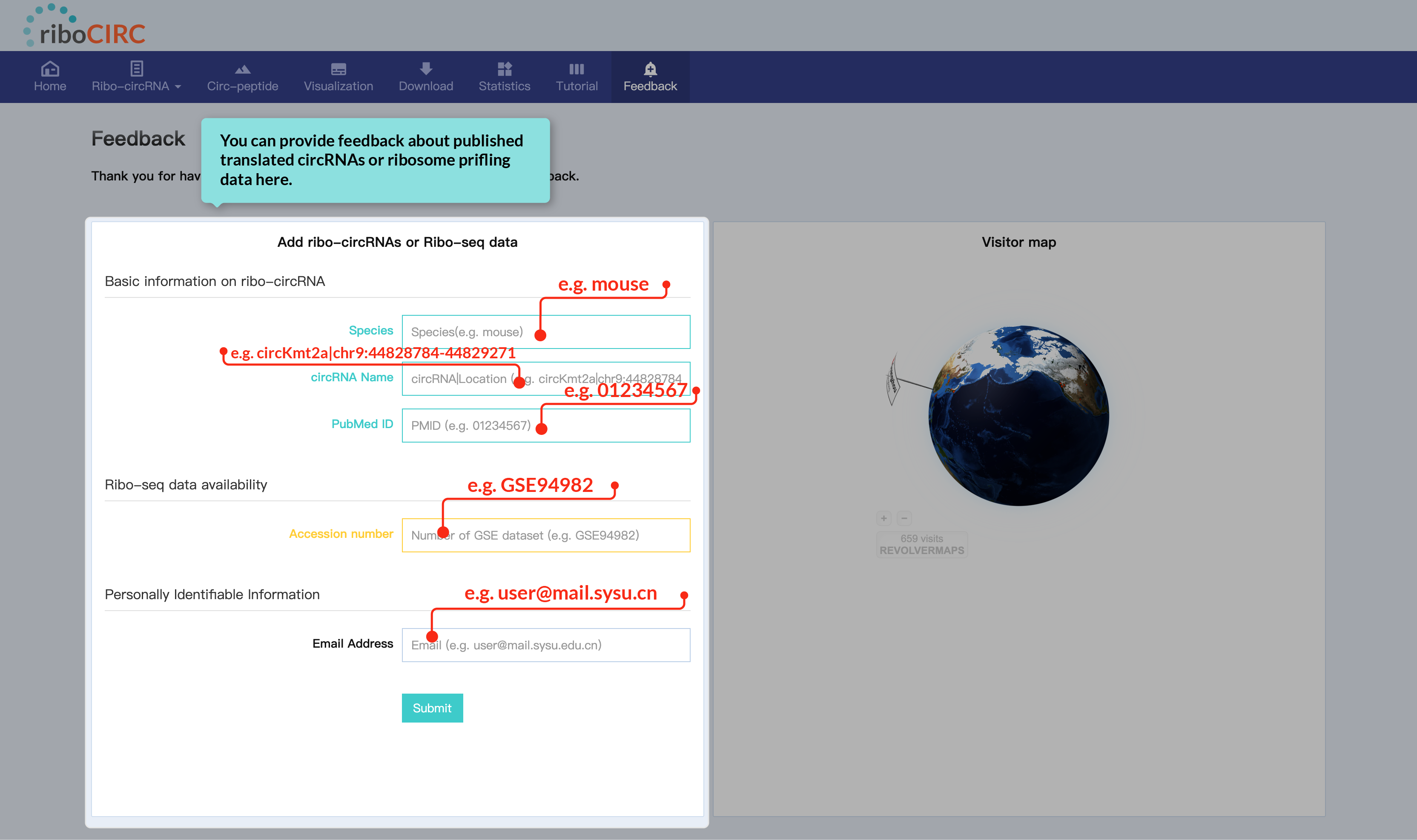
The Feedback page includes a dedicated form for comments on reported translatable circRNAs or ribosome profiling data. We welcome your specific and helpful suggestions.
0. Pipeline for identifying and annotating translatable circRNAs
1. CircRNA collection and processing
CircRNA data for six model organisms were compiled from five databases: circAtlas, CSCD, circBase, circRNADb, and CIRCpedia. To ensure data consistency, genomic coordinates were converted to 0-based indexing, and genome assemblies were harmonized using the liftOver tool (e.g., hg19 to hg38, mm9 to mm10). Initial filtering steps were applied to retain circRNAs satisfying the following criteria: (1) derivation solely from chromosomes; (2) defined strand information; (3) unambiguous gene origin annotation; and (4) availability of sequence information. To further exclude potential annotation artifacts, a stringent validation pipeline was implemented. GTF annotation files and genomic sequences were obtained from GENCODE and RefSeq for the corresponding genome versions. Exonic and intronic sequences were extracted from the GTF files and aligned against the circRNA sequences using BLAST in short-read mode (default minimum alignment length of 7 bp) with stringent parameters (100% identity, -perc_identity 100 -task blastn-short -dust no). CircRNAs were excluded if either of the following conditions was met: (1) the backsplice junction was directly flanked by intronic sequence; or (2) the exon/intron composition of the circRNA could not be unambiguously determined.
2. Characterization of translatable circRNAs
Two approaches were used to identify ribo-circRNAs: (1) Annotation-guided prediction, leveraging known circRNAs and Ribo-seq data, and (2) Context-specific prediction, performed de novo using paired RNA-seq and Ribo-seq data. Transcribed circRNAs were identified from RNA-seq data using CIRCexplorer3. Full-length circRNA sequences were reconstructed using the circRNAFull package. To identify ribosome-associated circRNAs, Ribo-seq data were first mapped to the linear reference genome using Tophat2. Unmapped reads were then re-mapped to a circRNA pseudo-reference using BWA. A circRNA was considered ribosome-associated if it satisfied all the following criteria: (1) at least two unique BSJ-spanning reads, (2) a minimum overlap of 3 nt between the Ribo-seq read and each side of the BSJ, (3) a Ribo-seq read length between 25 and 35 nt, and (4) the presence of a BSJ-spanning ORF initiated by an NTG start codon.
3. Prediction of circRNA-derived ORFs
For each circRNA, exonic sequences were extracted, and ORFs were predicted in silico. ORFs were required to have a minimum length of 63 nt, initiate with an NTG start codon, and span backsplice junction by at least 3 bases. Briefly, the ORF prediction process involved: (1) concatenating circRNA sequences 2 or 4 times based on divisibility by 3 to create extended linear sequences; (2) scanning for potential ORFs starting with NTG and ending with canonical stop codons (TGA, TAA, TAG) or lacking in-frame stop codons (designated as "infinite" ORFs); (3) filtering candidates to ensure nucleotide length divisible by 3, minimum 63 nucleotides, and spanning at least 3 bases across the junction; (4) For each reading frame (0, 1, 2), the longest finite ORF was selected, but in the absence of a finite ORF, the infinite ORF was selected; and (5) calculating their positions to generate ORF structure, and translating them to amino acid sequences.
4. Detection of cross-species conserved circRNAs
Orthologous genes were identified using the OMA Orthologs database. 50 bp sequences flanking the junction were extracted to represent the BSJ sequence (if the length is less than 100 bp, the sequence is clipped from the center of the circRNA). Subsequently, all circRNA BSJ sequences from one species were aligned against those from other species using BLAT, with parameters set to -minIdentity=30 -tileSize=6. Alignment entries were filtered to retain only those corresponding to orthologous genes and exhibiting a sequence identity exceeding 50%. The reciprocal best-hit pair with the highest mutual alignment identity was designated as the orthologous circRNA pair.
5. Annotation of IRES elements in circRNAs
IRES sequences were retrieved from the IRESbase database and aligned against circRNA sequences using BLAST in short-read mode, requiring a minimum alignment length of 10 bp and no mismatches. For each circRNA, the IRES sequence with the highest alignment score was selected as the final IRES elements.
6. Annotation of m6A sites in circRNAs
m6A modification peaks identified by three different peak-calling tools (exomePeak, MeTPeak, and MACS2) were obtained from the REPIC database. The genomic coordinates of these peaks were intersected with circRNA genomic coordinates to identify m6A modification sites. m6A sites were defined as adenine (A) positions within the "RAC" motif (where R represents any purine) located within the overlapping regions.
7. Annotation of RNA editing sites in circRNAs
RNA editing site coordinates were obtained from the REDIportal database and intersected with circRNA genomic coordinates. Overlapping sites were considered RNA editing sites corresponding to the circRNAs, with the requirement that gene annotations for the RNA editing site and the circRNA were consistent.
8. Annotation of RBP binding in circRNAs
RBP binding site coordinates were obtained from the POSTAR3 database and intersected with circRNA genomic coordinates. Only RBP binding sites completely contained within the circRNA coordinates were considered. Overlapping RBP binding sites were clustered using a 200 bp distance threshold, with each cluster potentially containing multiple RBPs.